Quest for the right Drug
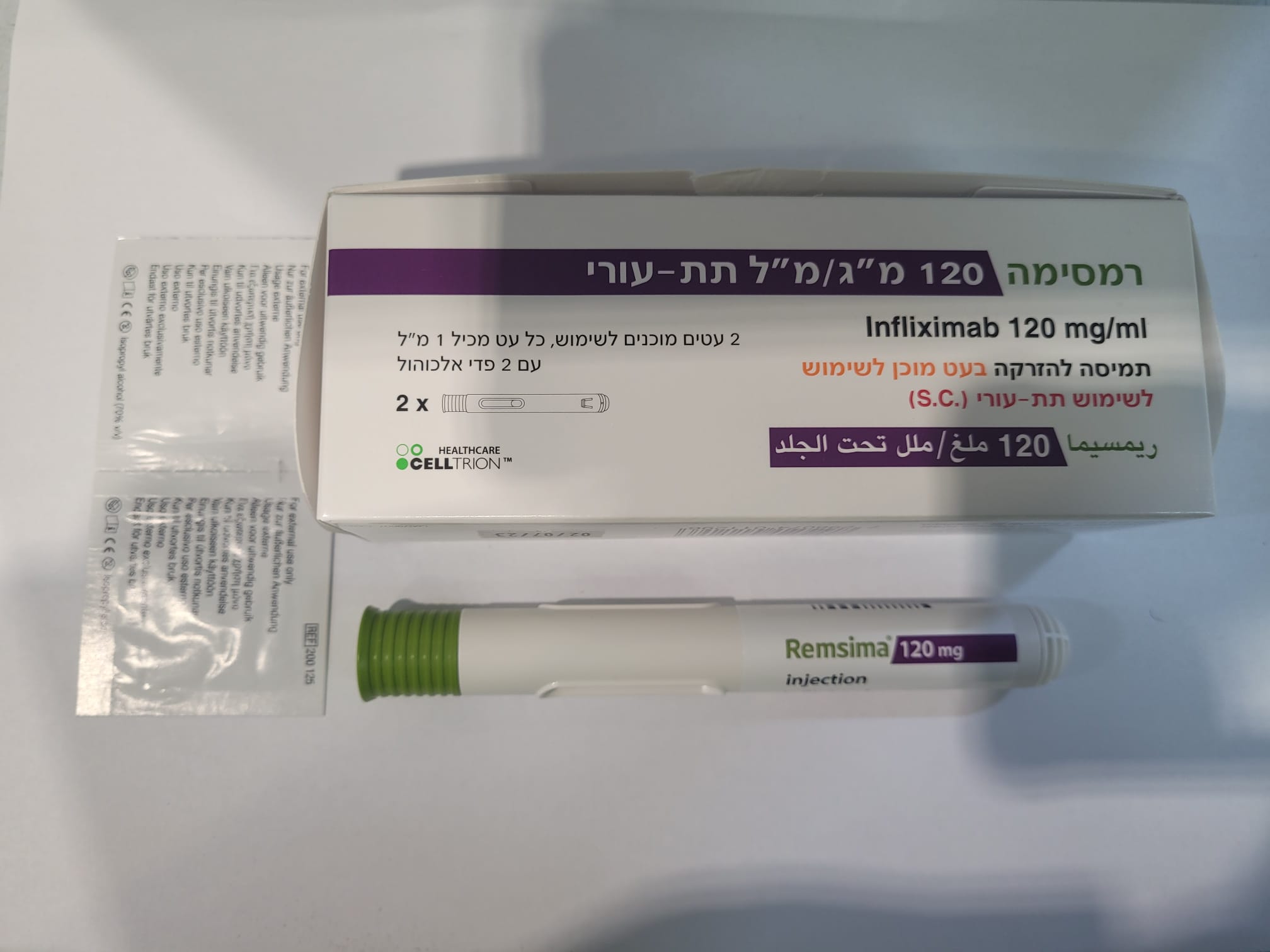
רמסימה 120 מ"ג/מ"ל תת-עורי REMSIMA 120 MG/ML S.C. (INFLIXIMAB)
תרופה במרשם
תרופה בסל
נרקוטיקה
ציטוטוקסיקה
צורת מתן:
תת-עורי : S.C
צורת מינון:
תמיסה להזרקה : SOLUTION FOR INJECTION
עלון לרופא
מינוניםPosology התוויות
Indications תופעות לוואי
Adverse reactions התוויות נגד
Contraindications אינטראקציות
Interactions מינון יתר
Overdose הריון/הנקה
Pregnancy & Lactation אוכלוסיות מיוחדות
Special populations תכונות פרמקולוגיות
Pharmacological properties מידע רוקחי
Pharmaceutical particulars אזהרת שימוש
Special Warning עלון לרופא
Physicians Leaflet
Pharmacological properties : תכונות פרמקולוגיות
Pharmacodynamic Properties
5.1 Pharmacodynamic properties Pharmacotherapeutic group: immunosuppressants, tumour necrosis factor alpha (TNFα) inhibitors, ATC code: L04AB02 Mechanism of action Infliximab is a chimeric human-murine monoclonal antibody that binds with high affinity to both soluble and transmembrane forms of TNFα but not to lymphotoxin α (TNF ). Pharmacodynamic effects Infliximab inhibits the functional activity of TNFα in a wide variety of in vitro bioassays. Infliximab prevented disease in transgenic mice that develop polyarthritis as a result of constitutive expression of human TNFα and when administered after disease onset, it allowed eroded joints to heal. In vivo, infliximab rapidly forms stable complexes with human TNFα, a process that parallels the loss of TNFα bioactivity. Elevated concentrations of TNFα have been found in the joints of rheumatoid arthritis patients and correlate with elevated disease activity. In rheumatoid arthritis, treatment with infliximab reduced infiltration of inflammatory cells into inflamed areas of the joint as well as expression of molecules mediating cellular adhesion, chemoattraction and tissue degradation. After infliximab treatment, patients exhibited decreased levels of serum interleukin 6 (IL-6) and C-reactive protein (CRP), and increased haemoglobin levels in rheumatoid arthritis patients with reduced haemoglobin levels, compared with baseline. Peripheral blood lymphocytes further showed no significant decrease in number or in proliferative responses to in vitro mitogenic stimulation when compared with untreated patients’ cells. In psoriasis patients, treatment with infliximab resulted in decreases in epidermal inflammation and normalisation of keratinocyte differentiation in psoriatic plaques. In psoriatic arthritis, short term treatment with infliximab reduced the number of T-cells and blood vessels in the synovium and psoriatic skin. Histological evaluation of colonic biopsies, obtained before and 4 weeks after administration of infliximab, revealed a substantial reduction in detectable TNFα. Infliximab treatment of Crohn’s disease patients was also associated with a substantial reduction of the commonly elevated serum inflammatory marker, CRP. Total peripheral white blood cell counts were minimally affected in infliximab-treated patients, although changes in lymphocytes, monocytes and neutrophils reflected shifts towards normal ranges. Peripheral blood mononuclear cells (PBMC) from infliximab-treated patients showed undiminished proliferative responsiveness to stimuli compared with untreated patients, and no substantial changes in cytokine production by stimulated PBMC were observed following treatment with infliximab. Analysis of lamina propria mononuclear cells obtained by biopsy of the intestinal mucosa showed that infliximab treatment caused a reduction in the number of cells capable of expressing TNFα and interferon . Additional histological studies provided evidence that treatment with infliximab reduces the infiltration of inflammatory cells into affected areas of the intestine and the presence of inflammation markers at these sites. Endoscopic studies of intestinal mucosa have shown evidence of mucosal healing in infliximab-treated patients. Clinical efficacy and safety Adult rheumatoid arthritis Intravenous formulation The efficacy of infliximab intravenous formulation was assessed in two multicentre, randomised, double-blind, pivotal clinical studies: ATTRACT and ASPIRE. In both studies concurrent use of stable doses of folic acid, oral corticosteroids (≤10 mg/day) and/or non-steroidal anti-inflammatory drugs (NSAIDs) was permitted. The primary endpoints were the reduction of signs and symptoms as assessed by the ACR criteria (ACR20 for ATTRACT, landmark ACR-N for ASPIRE), the prevention of structural joint damage, and the improvement in physical function. A reduction in signs and symptoms was defined to be at least a 20% improvement (ACR20) in both tender and swollen joint counts, and in 3 of the following 5 criteria: (1) evaluator’s global assessment, (2) patient’s global assessment, (3) functional/disability measure, (4) visual analogue pain scale and (5) erythrocyte sedimentation rate or C-reactive protein. ACR-N uses the same criteria as the ACR20, calculated by taking the lowest percent improvement in swollen joint count, tender joint count, and the median of the remaining 5 components of the ACR response. Structural joint damage (erosions and joint space narrowing) in both hands and feet was measured by the change from baseline in the total van der Heijde-modified Sharp score (0-440). The Health Assessment Questionnaire (HAQ; scale 0-3) was used to measure patients’ average change from baseline scores over time, in physical function. The ATTRACT study evaluated responses at 30, 54 and 102 weeks in a placebo-controlled study of 428 patients with active rheumatoid arthritis despite treatment with methotrexate. Approximately 50% of patients were in functional Class III. Patients received placebo, 3 mg/kg or 10 mg/kg infliximab at weeks 0, 2 and 6, and then every 4 or 8 weeks thereafter. All patients were on stable methotrexate doses (median 15 mg/wk) for 6 months prior to enrolment and were to remain on stable doses throughout the study. Results from week 54 (ACR20, total van der Heijde-modified Sharp score and HAQ) are shown in Table 3. Higher degrees of clinical response (ACR50 and ACR70) were observed in all infliximab groups at 30 and 54 weeks compared with methotrexate alone. A reduction in the rate of the progression of structural joint damage (erosions and joint space narrowing) was observed in all infliximab groups at 54 weeks (Table 3). The effects observed at 54 weeks were maintained through 102 weeks. Due to a number of treatment withdrawals, the magnitude of the effect difference between infliximab and the methotrexate alone group cannot be defined. Table 3 Effects on ACR20, Structural Joint Damage and Physical Function at week 54, ATTRACT Infliximabb a Control 3 mg/kg 3 mg/kg 10 mg/kg 10 mg/kg All q 8 wks q 4 wks q 8 wks q 4 wks infliximabb Patients with ACR20 15/88 36/86 41/86 51/87 48/81 176/340 response/Patients (17%) (42%) (48%) (59%) (59%) (52%) evaluated (%) Total scored (van der Heijde-modified Sharp score) Change from baseline 7.0±10.3 1.3 ± 6.0 1.6 ± 8.5 0.2 ± 3.6 -0.7 ± 3.8 0.6 ± 5.9 (Mean ± SDc) Median 4.0 0.5 0.1 0.5 -0.5 0.0 (Interquartile range) (0.5,9.7) (-1.5,3.0) (-2.5,3.0) (-1.5,2.0) (-3.0,1.5) (-1.8,2.0) Patients with no 13/64 34/71 35/71 37/77 44/66 150/285 deterioration/patients (20%) (48%) (49%) (48%) (67%) (53%) evaluated (%)c HAQ change from 87 86 85 87 81 339 baseline over timee (patients evaluated) Mean ± SDc 0.2 ± 0.3 0.4 ± 0.3 0.5 ± 0.4 0.5 ± 0.5 0.4 ± 0.4 0.4 ± 0.4 a control = All patients had active RA despite treatment with stable methotrexate doses for 6 months prior to enrolment and were to remain on stable doses throughout the study. Concurrent use of stable doses of oral corticosteroids (≤10 mg/day) and/or NSAIDs was permitted, and folate supplementation was given. b all infliximab doses given in combination with methotrexate and folate with some on corticosteroids and/or NSAIDs c p <0.001, for each infliximab treatment group vs. control d greater values indicate more joint damage. e HAQ = Health Assessment Questionnaire; greater values indicate less disability. The ASPIRE study evaluated responses at 54 weeks in 1,004 methotrexate naive patients with early (≤3 years disease duration, median 0.6 years) active rheumatoid arthritis (median swollen and tender joint count of 19 and 31, respectively). All patients received methotrexate (optimised to 20 mg/wk by week 8) and either placebo, 3 mg/kg or 6 mg/kg infliximab at weeks 0, 2, and 6 and every 8 weeks thereafter. Results from week 54 are shown in Table 4. After 54 weeks of treatment, both doses of infliximab + methotrexate resulted in statistically significantly greater improvement in signs and symptoms compared to methotrexate alone as measured by the proportion of patients achieving ACR20, 50 and 70 responses. In ASPIRE, more than 90% of patients had at least two evaluable X-rays. Reduction in the rate of progression of structural damage was observed at weeks 30 and 54 in the infliximab + methotrexate groups compared to methotrexate alone. Table 4 Effects on ACRn, Structural Joint Damage and Physical Function at week 54, ASPIRE Infliximab + MTX Placebo 3 mg/kg 6 mg/kg Combined + MTX Subjects randomised 282 359 363 722 Percentage ACR improvement Mean ± SDa 24.8 ± 59.7 37.3 ± 52.8 42.0 ± 47.3 39.6 ± 50.1 Change from baseline in total van der Heijde-modified Sharp scoreb Mean ± SDa 3.70±9.61 0.42±5.82 0.51±5.55 0.46±5.68 Median 0.43 0.00 0.00 0.00 Improvement from baseline in HAQ averaged over time from week 30 to week 54c Mean ± SDd 0.68 ± 0.63 0.80 ± 0.65 0.88 ± 0.65 0.84 ± 0.65 a p <0.001, for each infliximab treatment group vs control b greater values indicate more joint damage. c HAQ = Health Assessment Questionnaire; greater values indicate less disability. d p = 0.030 and <0.001 for the 3 mg/kg and 6 mg/kg treatment groups respectively vs. placebo + MTX. Data to support dose titration in rheumatoid arthritis come from ATTRACT, ASPIRE and the START study. START was a randomised, multicentre, double-blind, 3-arm, parallel-group safety study. In one of the study arms (group 2, n=329), patients with an inadequate response were allowed to dose titrate with 1.5 mg/kg increments from 3 up to 9 mg/kg. The majority (67%) of these patients did not require any dose titration. Of the patients who required a dose titration, 80% achieved clinical response and the majority (64%) of these required only one adjustment of 1.5 mg/kg. Subcutaneous formulation The efficacy of subcutaneous infliximab in rheumatoid arthritis patients was assessed in a randomised, parallel-group pivotal Phase I/III study consisting of two parts: Part 1 to determine the optimal dose of subcutaneous infliximab and Part 2 to demonstrate non-inferiority in terms of efficacy of subcutaneous infliximab compared to intravenous infliximab treatment in a double-blind setting. In Part 2 of this study, among 357 patients who were enrolled to receive 2 doses of Remsima 3 mg/kg intravenously at Weeks 0 and 2, 167 patients were randomised to receive Remsima 120 mg/ml S.C. at Week 6 and every 2 weeks up to Week 54, while 176 patients were randomised to receive Remsima 3 mg/kg intravenously at Weeks 6, 14 and 22 and then switched to Remsima 120 mg/ml S.C. at Week 30 once-every 2 weeks up to Week 54. Methotrexate was given concomitantly. The primary endpoint of the study was the treatment difference of the change from baseline of DAS28 (CRP) at Week 22. The estimate of treatment difference was 0.27 with corresponding lower limit of the two-sided 95% confidence interval [CI] of 0.02 (95% CI: 0.02, 0.52), which was greater than the pre-specified non-inferiority margin of -0.6 indicating non-inferiority of Remsima subcutaneous formulation to Remsima intravenous formulation. The analysis of other efficacy endpoints showed that efficacy profile of Remsima subcutaneous formulation compared to Remsima intravenous formulation in RA patients was generally comparable in terms of disease activity measured by DAS28 (CRP and ESR) and ACR response up to Week 54. The mean scores for DAS28 (CRP) and DAS28 (ESR) gradually decreased from baseline at each time point until Week 54 in each treatment arm (see Table 5 and Table 6, respectively). Table 5 Mean (SD) Actual Values of DAS28 (CRP and ESR)___________________ DAS28 (CRP) DAS28 (ESR) Remsima IV Remsima SC Remsima IV Remsima SC 3 mg/kgb 120 mg 3 mg/kgb 120 mg Visit (N=174) (N=165) (N=174) (N=165) Baseline 5.9 (0.8) 6.0 (0.8) 6.6 (0.8) 6.7 (0.8) Week 6 4.1 (1.2) 4.0 (1.2) 4.8 (1.3) 4.6 (1.2) Week 22 3.5 (1.2)a 3.3 (1.1)a 4.1 (1.3) 4.0 (1.1) Week 54 2.9 (1.2)b 2.8 (1.1) 3.4 (1.3)b 3.4 (1.2) a Two-sided 95% CI for difference in the mean change from baseline for DAS28 (CRP) at Week 22 was well above the pre-defined non-inferiority margin of -0.6 b Remsima IV was switched to Remsima SC at Week 30 Table 6 Proportions of Patients Achieving Clinical Response According to the ACR Criteria_ ACR20 ACR50 ACR70 Remsima IV Remsima SC Remsima IV Remsima SC Remsima IV Remsima SC 3 mg/kga 120 mg 3 mg/kga 120 mg 3 mg/kga 120 mg Visit (N=174) (N=165) (N=174) (N=165) (N=174) (N=165) Week 6 103 (59.2%) 107 (64.8%) 45 (25.9%) 47 (28.5%) 18 (10.3%) 19 (11.5%) Week 22 137 (78.7%) 139 (84.2%) 90 (51.7%) 85 (51.5%) 49 (28.2%) 46 (27.9%) Week 54 125 (71.8%)a 132 (80.0%) 101 (58.0%) a 108 (65.5%) 68 (39.1%) a 77 (46.7%) a Remsima IV was switched to Remsima SC at Week 30 Adult Crohn’s disease Intravenous formulation Induction treatment in moderately to severely active Crohn’s disease The efficacy of a single dose treatment with infliximab intravenous formulation was assessed in 108 patients with active Crohn’s disease (CDAI ≥220 ≤400) in a randomised, double-blinded, placebo-controlled, dose-response study. Of these 108 patients, 27 were treated with the recommended dosage of infliximab 5 mg/kg. All patients had experienced an inadequate response to prior conventional therapies. Concurrent use of stable doses of conventional therapies was permitted, and 92% of patients continued to receive these therapies. The primary endpoint was the proportion of patients who experienced a clinical response, defined as a decrease in CDAI by ≥70 points from baseline at the 4-week evaluation and without an increase in the use of medicinal products or surgery for Crohn’s disease. Patients who responded at week 4 were followed to week 12. Secondary endpoints included the proportion of patients in clinical remission at week 4 (CDAI <150) and clinical response over time. At week 4, following administration of a single dose, 22/27 (81%) of infliximab-treated patients receiving a 5 mg/kg dose achieved a clinical response vs. 4/25 (16%) of the placebo-treated patients (p <0.001). Also at week 4, 13/27 (48%) of infliximab-treated patients achieved a clinical remission (CDAI <150) vs. 1/25 (4%) of placebo-treated patients. A response was observed within 2 weeks, with a maximum response at 4 weeks. At the last observation at 12 weeks, 13/27 (48%) of infliximab-treated patients were still responding. Maintenance treatment in moderately to severely active Crohn’s disease in adults The efficacy of repeated infusions with intravenous infliximab was studied in a 1-year clinical study (ACCENT I). A total of 573 patients with moderately to severely active Crohn’s disease (CDAI ≥220 ≤400) received a single infusion of 5 mg/kg at week 0. 178 of the 580 enrolled patients (30.7%) were defined as having severe disease (CDAI score > 300 and concomitant corticosteroid and/or immunosuppressants) corresponding to the population defined in the indication (see section 4.1). At week 2, all patients were assessed for clinical response and randomised to one of 3 treatment groups; a placebo maintenance group, 5 mg/kg maintenance group and 10 mg/kg maintenance group. All 3 groups received repeated infusions at week 2, 6 and every 8 weeks thereafter. Of the 573 patients randomised, 335 (58%) achieved clinical response by week 2. These patients were classified as week-2 responders and were included in the primary analysis (see Table 7). Among patients classified as non-responders at week 2, 32% (26/81) in the placebo maintenance group and 42% (68/163) in the infliximab group achieved clinical response by week 6. There was no difference between groups in the number of late responders thereafter. The co-primary endpoints were the proportion of patients in clinical remission (CDAI <150) at week 30 and time to loss of response through week 54. Corticosteroid tapering was permitted after week 6. Table 7 Effects on response and remission rate, data from ACCENT I (Week-2 responders) ACCENT I (Week-2 responders) % of Patients Placebo Infliximab Infliximab Maintenance Maintenance Maintenance 5 mg/kg 10 mg/kg (n=110) (n=113) (n=112) (p value) (p value) Median time to loss of response 19 weeks 38 weeks >54 weeks through week 54 (0.002) (<0.001) Week 30 Clinical Responsea 27.3 51.3 59.1 (<0.001) (<0.001) Clinical Remission 20.9 38.9 45.5 (0.003) (<0.001) Steroid-Free Remission 10.7 (6/56) 31.0 (18/58) 36.8 (21/57) (0.008) (0.001) Week 54 Clinical Responsea 15.5 38.1 47.7 (<0.001) (<0.001) Clinical Remission 13.6 28.3 38.4 (0.007) (<0.001) Sustained Steroid-Free 5.7 (3/53) 17.9 (10/56) 28.6 (16/56) Remissionb (0.075) (0.002) a Reduction in CDAI ≥25% and ≥70 points. b CDAI <150 at both Week 30 and 54 and not receiving corticosteroids in the 3 months prior to Week 54 among patients who were receiving corticosteroids at baseline. Beginning at week 14, patients who had responded to treatment, but subsequently lost their clinical benefit, were allowed to cross over to a dose of infliximab 5 mg/kg higher than the dose to which they were originally randomised. Eighty nine percent (50/56) of patients who lost clinical response on infliximab 5 mg/kg maintenance therapy after week 14 responded to treatment with infliximab 10 mg/kg. Improvements in quality of life measures, a reduction in disease-related hospitalisations and corticosteroid use were seen in the infliximab maintenance groups compared with the placebo maintenance group at weeks 30 and 54. Infliximab with or without AZA was assessed in a randomised, double-blind, active comparator study (SONIC) of 508 adult patients with moderate to severe Crohn’s disease (CDAI ≥220 ≤450) who were naive to biologics and immunosuppressants and had a median disease duration of 2.3 years. At baseline 27.4% of patients were receiving systemic corticosteroids, 14.2% of patients were receiving budesonide, and 54.3% of patients were receiving 5-ASA compounds. Patients were randomised to receive AZA monotherapy, infliximab monotherapy, or infliximab plus AZA combination therapy. Infliximab was administered at a dose of 5 mg/kg at weeks 0, 2, 6, and then every 8 weeks. AZA was given at a dose of 2.5 mg/kg daily. The primary endpoint of the study was corticosteroid-free clinical remission at week 26, defined as patients in clinical remission (CDAI of <150) who, for at least 3 weeks, had not taken oral systemic corticosteroids (prednisone or equivalent) or budesonide at a dose >6 mg/day. For results see Table 8. The proportions of patients with mucosal healing at week 26 were significantly greater in the infliximab plus AZA combination (43.9%, p<0.001) and infliximab monotherapy groups (30.1%, p=0.023) compared to the AZA monotherapy group (16.5%). Table 8 Percent of patients achieving corticosteroid-free clinical remission at Week 26, SONIC AZA Infliximab Infliximab + AZA Monotherapy Monotherapy Combination therapy Week 26 All randomised patients 30.0% 44.4% (75/169) 56.8% (96/169) (51/170) (p=0.006)* (p<0.001)* * p-values represent each infliximab treatment group vs. AZA monotherapy. Similar trends in the achievement of corticosteroid-free clinical remission were observed at week 50. Furthermore, improved quality of life as measured by IBDQ was observed with infliximab. Induction treatment in fistulising active Crohn’s disease The efficacy was assessed in a randomised, double-blinded, placebo-controlled study in 94 patients with fistulising Crohn’s disease who had fistulae that were of at least 3 months’ duration. Thirty one of these patients were treated with infliximab intravenous formulation 5 mg/kg. Approximately 93% of the patients had previously received antibiotic or immunosuppressive therapy. Concurrent use of stable doses of conventional therapies was permitted, and 83% of patients continued to receive at least one of these therapies. Patients received three doses of either placebo or infliximab at weeks 0, 2 and 6. Patients were followed up to 26 weeks. The primary endpoint was the proportion of patients who experienced a clinical response, defined as ≥50% reduction from baseline in the number of fistulae draining upon gentle compression on at least two consecutive visits (4 weeks apart), without an increase in the use of medicinal products or surgery for Crohn’s disease. Sixty eight percent (21/31) of infliximab-treated patients receiving a 5 mg/kg dose regimen achieved a clinical response vs. 26% (8/31) placebo-treated patients (p=0.002). The median time to onset of response in the infliximab-treated group was 2 weeks. The median duration of response was 12 weeks. Additionally, closure of all fistulae was achieved in 55% of infliximab-treated patients compared with 13% of placebo-treated patients (p=0.001). Maintenance treatment in fistulising active Crohn’s disease The efficacy of repeated infusions with infliximab in patients with fistulising Crohn’s disease was studied in a 1-year clinical study (ACCENT II). A total of 306 patients received 3 doses of intravenous infliximab 5 mg/kg at week 0, 2 and 6. At baseline, 87% of the patients had perianal fistulae, 14% had abdominal fistulae, 9% had rectovaginal fistulae. The median CDAI score was 180. At week 14, 282 patients were assessed for clinical response and randomised to receive either placebo or 5 mg/kg infliximab every 8 weeks through week 46. Week-14 responders (195/282) were analysed for the primary endpoint, which was time from randomisation to loss of response (see Table 9). Corticosteroid tapering was permitted after week 6. Table 9 Effects on response rate, data from ACCENT II (Week-14 responders) ACCENT II (Week-14 responders) Placebo Infliximab p-value Maintenance Maintenance (n=99) (5 mg/kg) (n=96) Median time to loss of response 14 weeks >40 weeks <0.001 through week 54 Week 54 Fistula Response (%)a 23.5 46.2 0.001 Complete fistula response (%)b 19.4 36.3 0.009 a A ≥50% reduction from baseline in the number of draining fistulas over a period of ≥4 weeks. b Absence of any draining fistulas. Beginning at week 22, patients who initially responded to treatment and subsequently lost their response were eligible to cross over to active re-treatment every 8 weeks at a dose of infliximab 5 mg/kg higher than the dose to which they were originally randomised. Among patients in the infliximab 5 mg/kg group who crossed over because of loss of fistula response after week 22, 57% (12/21) responded to re-treatment with infliximab 10 mg/kg every 8 weeks. There was no significant difference between placebo and infliximab for the proportion of patients with sustained closure of all fistulas through week 54, for symptoms such as proctalgia, abscesses and urinary tract infection or for number of newly developed fistulas during treatment. Maintenance therapy with infliximab every 8 weeks significantly reduced disease-related hospitalisations and surgeries compared with placebo. Furthermore, a reduction in corticosteroid use and improvements in quality of life were observed. Subcutaneous formulation The efficacy of subcutaneous infliximab in active Crohn’s disease and active ulcerative colitis patients was assessed in an open-label, randomised, parallel-group, Phase I study consisting of two parts: Part 1 to determine the optimal dose of subcutaneous infliximab and Part 2 to demonstrate non-inferiority in terms of PK of subcutaneous infliximab compared to intravenous infliximab treatment. In Part 1 of this study, 45 patients with active Crohn’s disease were enrolled to receive 2 doses of Remsima 5 mg/kg intravenously at Weeks 0 and 2 and subsequently 44 patients were randomised into four cohorts to receive Remsima 5 mg/kg intravenously (n=13) at Week 6 and every 8 weeks up to Week 54, Remsima 120 mg subcutaneously (n=11), Remsima 180 mg subcutaneously (n=12) or Remsima 240 mg subcutaneously (n=8) at Week 6 and every 2 weeks up to Week 54. In Part 2 of this study, among 136 patients (57 patients with active Crohn’s disease and 79 patients with active ulcerative colitis) who were enrolled to receive 2 doses of Remsima 5 mg/kg intravenously at Weeks 0 and 2, 66 patients (28 patients with active Crohn’s disease and 38 patients with active ulcerative colitis) were randomised to receive Remsima 120/ 240 mg subcutaneously at Week 6 and every 2 weeks up to Week 54, while 65 patients (25 patients with active Crohn’s disease and 40 patients with active ulcerative colitis) were randomised to receive Remsima 5 mg/kg intravenously at Week 6, 14 and 22 and then switched to Remsima 120/ 240 mg subcutaneous formulation at Week 30 once-every 2 weeks up to Week 54. The dosage of Remsima 120/ 240 mg subcutaneous formulation was determined based on the patient’s body weight at Week 6 for those who received Remsima subcutaneously and at Week 30 for those who switched to Remsima subcutaneous formulation (Remsima subcutaneous 120 mg for patients <80 kg; 240 mg for patients ≥80 kg). In active Crohn’s disease patients, the descriptive efficacy results following Remsima 120 mg subcutaneous formulation were generally comparable to Remsima 5 mg/kg intravenous formulation in terms of clinical response (CDAI-70 response defined as a decrease in CDAI by ≥70 points and CDAI-100 response defined as ≥100 points from baseline), clinical remission (defined as an absolute CDAI score of <150 points) and endoscopy assessments (endoscopic response defined as a decrease in ≥50% of overall Simplified Endoscopic Activity Score for Crohn’s Disease (SES-CD) score from the baseline value and endoscopic remission defined as an absolute SES-CD score of ≤2 points). Adult ulcerative colitis Intravenous formulation The safety and efficacy of intravenous infliximab were assessed in two (ACT 1 and ACT 2) randomised, double-blind, placebo-controlled clinical studies in adult patients with moderately to severely active ulcerative colitis (Mayo score 6 to 12; Endoscopy subscore ≥ 2) with an inadequate response to conventional therapies [oral corticosteroids, aminosalicylates and/or immunomodulators (6-MP, AZA)]. Concomitant stable doses of oral aminosalicylates, corticosteroids, and/or immunomodulatory agents were permitted. In both studies, patients were randomised to receive either placebo, 5 mg/kg infliximab, or 10 mg/kg infliximab at weeks 0, 2, 6, 14 and 22, and in ACT 1 at weeks 30, 38 and 46. Corticosteroid taper was permitted after week 8. Table 10 Effects on clinical response, clinical remission and mucosal healing at Weeks 8 and 30. Combined data from ACT 1 & 2 Infliximab Placebo 5 mg/kg 10 mg/kg Combined Subjects randomised 244 242 242 484 Percentage of subjects in clinical response and in sustained clinical response Clinical response at Week 8a 33.2% 66.9% 65.3% 66.1% Clinical response at Week 30a 27.9% 49.6% 55.4% 52.5% Sustained response (clinical response at both Week 8 and 19.3% 45.0% 49.6% 47.3% Week 30)a Percentage of subjects in clinical remission and sustained remission Clinical remission at Week 8a 10.2% 36.4% 29.8% 33.1% Clinical remission at Week 30a 13.1% 29.8% 36.4% 33.1% Sustained remission(in remission at both Week 8 and Week 30)a 5.3% 19.0% 24.4% 21.7% Infliximab Placebo 5 mg/kg 10 mg/kg Combined Percentage of subjects with mucosal healing Mucosal healing at Week 8a 32.4% 61.2% 60.3% 60.7% Mucosal healing at Week 30a 27.5% 48.3% 52.9% 50.6% a p <0.001, for each infliximab treatment group vs. placebo. The efficacy of infliximab through week 54 was assessed in the ACT 1 study. At 54 weeks, 44.9% of patients in the combined infliximab treatment group were in clinical response compared to 19.8% in the placebo treatment group (p<0.001). Clinical remission and mucosal healing occurred in a greater proportion of patients in the combined infliximab treatment group compared to the placebo treatment group at week 54 (34.6% vs. 16.5%, p<0.001 and 46.1% vs. 18.2%, p<0.001, respectively). The proportions of patients in sustained response and sustained remission at week 54 were greater in the combined infliximab treatment group than in the placebo treatment group (37.9% vs. 14.0%, p<0.001; and 20.2% vs. 6.6%, p<0.001, respectively). A greater proportion of patients in the combined infliximab treatment group were able to discontinue corticosteroids while remaining in clinical remission compared to the placebo treatment group at both week 30 (22.3% vs. 7.2%, p <0.001, pooled ACT 1 & ACT 2 data) and week 54 (21.0% vs. 8.9%, p=0.022, ACT 1 data). The pooled data analysis from the ACT 1 and ACT 2 studies and their extensions, analysed from baseline through 54 weeks, demonstrated a reduction of ulcerative colitis-related hospitalisations and surgical procedures with infliximab treatment. The number of ulcerative colitis-related hospitalisations was significantly lower in the 5 and 10 mg/kg infliximab treatment groups than in the placebo group (mean number of hospitalisations per 100 subject-years: 21 and 19 vs. 40 in the placebo group; p=0.019 and p=0.007, respectively). The number of ulcerative colitis-related surgical procedures was also lower in the 5 and 10 mg/kg infliximab treatment groups than in the placebo group (mean number of surgical procedures per 100 subject-years: 22 and 19 vs. 34; p=0.145 and p=0.022, respectively). The proportion of subjects who underwent colectomy at any time within 54 weeks following the first infusion of study agent were collected and pooled from the ACT 1 and ACT 2 studies and their extensions. Fewer subjects underwent colectomy in the 5 mg/kg infliximab group (28/242 or 11.6% [N.S.]) and the 10 mg/kg infliximab group (18/242 or 7.4% [p=0.011]) than in the placebo group (36/244; 14.8%). The reduction in incidence of colectomy was also examined in another randomised, double-blind study (C0168Y06) in hospitalised patients (n=45) with moderately to severely active ulcerative colitis who failed to respond to intravenous corticosteroids and who were therefore at higher risk for colectomy. Significantly fewer colectomies occurred within 3 months of study infusion in patients who received a single dose of 5 mg/kg infliximab compared to patients who received placebo (29.2% vs. 66.7% respectively, p=0.017). In ACT 1 and ACT 2, infliximab improved quality of life, confirmed by statistically significant improvement in both a disease specific measure, IBDQ, and by improvement in the generic 36-item short form survey SF-36. Subcutaneous formulation The efficacy of subcutaneous infliximab in active ulcerative colitis patients was assessed in Part 2 of an open-label, randomised, parallel-group, Phase I study. For study details, see Section 5.1 on Crohn’s disease, subcutaneous formulation. In active ulcerative colitis patients, the descriptive efficacy results following Remsima 120 mg subcutaneous formulation were generally comparable to Remsima 5 mg/kg intravenous formulation in terms of clinical response (defined as a decrease from baseline in total Mayo score of at least 3 points and at least 30% or a decrease from baseline in partial Mayo score at least 2 points, with an accompanying decrease from baseline in the subscore for rectal bleeding of at least 1 point, or an absolute subscore for rectal bleeding of 0 or 1), clinical remission (defined as a total Mayo score of ≤2 points with no individual subscore exceeding 1 point, or partial Mayo score of ≤1 point) and mucosal healing (defined as absolute endoscopic subscore of 0 or 1 from Mayo Scoring System). Adult ankylosing spondylitis Intravenous formulation Efficacy and safety of infliximab intravenous formulation were assessed in two multicentre, double-blind, placebo-controlled studies in patients with active ankylosing spondylitis (Bath Ankylosing Spondylitis Disease Activity Index [BASDAI] score ≥ 4 and spinal pain ≥ 4 on a scale of 1-10). In the first study (P01522), which had a 3-month double-blind phase, 70 patients received either 5 mg/kg infliximab or placebo at weeks 0, 2, 6 (35 patients in each group). At week 12, placebo patients were switched to infliximab 5 mg/kg every 6 weeks up to week 54. After the first year of the study, 53 patients continued into an open-label extension to week 102. In the second clinical study (ASSERT), 279 patients were randomised to receive either placebo (Group 1, n=78) or 5 mg/kg infliximab (Group 2, n=201) at 0, 2 and 6 weeks and every 6 weeks to week 24. Thereafter, all subjects continued on infliximab every 6 weeks to week 96. Group 1 received 5 mg/kg infliximab. In Group 2, starting with the week 36 infusion, patients who had a BASDAI ≥3 at 2 consecutive visits, received 7.5 mg/kg infliximab every 6 weeks thereafter through week 96. In ASSERT, improvement in signs and symptoms was observed as early as week 2. At week 24, the number of ASAS 20 responders was 15/78 (19%) in the placebo group, and 123/201 (61%) in the 5 mg/kg infliximab group (p<0.001). There were 95 subjects from group 2 who continued on 5 mg/kg every 6 weeks. At 102 weeks there were 80 subjects still on infliximab treatment and among those, 71 (89%) were ASAS 20 responders. In P01522, improvement in signs and symptoms was also observed as early as week 2. At week 12, the number of BASDAI 50 responders were 3/35 (9%) in the placebo group, and 20/35 (57%) in the 5 mg/kg group (p<0.01). There were 53 subjects who continued on 5 mg/kg every 6 weeks. At 102 weeks there were 49 subjects still on infliximab treatment and among those, 30 (61%) were BASDAI 50 responders. In both studies, physical function and quality of life as measured by the BASFI and the physical component score of the SF-36 were also improved significantly. Adult psoriatic arthritis Intravenous formulation Efficacy and safety of infliximab intravenous formulation were assessed in two multicentre, double-blind, placebo-controlled studies in patients with active psoriatic arthritis. In the first clinical study (IMPACT), efficacy and safety of infliximab were studied in 104 patients with active polyarticular psoriatic arthritis. During the 16-week double-blind phase, patients received either 5 mg/kg infliximab or placebo at weeks 0, 2, 6, and 14 (52 patients in each group). Starting at week 16, placebo patients were switched to infliximab and all patients subsequently received 5 mg/kg infliximab every 8 weeks up to week 46. After the first year of the study, 78 patients continued into an open-label extension to week 98. In the second clinical study (IMPACT 2), efficacy and safety of infliximab were studied in 200 patients with active psoriatic arthritis (≥5 swollen joints and ≥5 tender joints). Forty six percent of patients continued on stable doses of methotrexate (≤25 mg/week). During the 24-week double-blind phase, patients received either 5 mg/kg infliximab or placebo at weeks 0, 2, 6, 14, and 22 (100 patients in each group). At week 16, 47 placebo patients with <10% improvement from baseline in both swollen and tender joint counts were switched to infliximab induction (early escape). At week 24, all placebo-treated patients crossed over to infliximab induction. Dosing continued for all patients through week 46. Key efficacy results for IMPACT and IMPACT 2 are shown in Table 11 below: Table 11 Effects on ACR and PASI in IMPACT and IMPACT 2 IMPACT IMPACT 2* Placebo Infliximab Infliximab Placebo Infliximab Infliximab (Week 16) (Week 16) (Week 98) (Week 24) (Week 24) (Week 54) Patients 52 52 N/Aa 100 100 100 randomised ACR response (% of patients) N 52 52 78 100 100 100 ACR 20 5 (10%) 34 (65%) 48 (62%) 16 (16%) 54 (54%) 53 (53%) response* ACR 50 0 (0%) 24 (46%) 35 (45%) 4 (4%) 41 (41%) 33 (33%) response* ACR 70 0 (0%) 15 (29%) 27 (35%) 2 (2%) 27 (27%) 20 (20%) response* PASI response (% of patients)b 87 83 82 N PASI 75 1 (1%) 50 (60%) 40 (48.8%) response** * ITT-analysis where subjects with missing data were included as non-responders. a Week 98 data for IMPACT includes combined placebo crossover and infliximab patients who entered the open-label extension. b Based on patients with PASI >2.5 at baseline for IMPACT, and patients with >3% BSA psoriasis skin involvement at baseline in IMPACT 2. ** PASI 75 response for IMPACT not included due to low N; p<0.001 for infliximab vs. placebo at week 24 for IMPACT 2. In IMPACT and IMPACT 2, clinical responses were observed as early as week 2 and were maintained through week 98 and week 54, respectively. Efficacy has been demonstrated with or without concomitant use of methotrexate. Decreases in parameters of peripheral activity characteristic of psoriatic arthritis (such as number of swollen joints, number of painful/tender joints, dactylitis and presence of enthesopathy) were seen in the infliximab-treated patients. Radiographic changes were assessed in IMPACT 2. Radiographs of hands and feet were collected at baseline, weeks 24 and 54. Infliximab treatment reduced the rate of progression of peripheral joint damage compared with placebo treatment at the week 24 primary endpoint as measured by change from baseline in total modified vdH-S score (mean ± SD score was 0.82 ± 2.62 in the placebo group compared with -0.70 ± 2.53 in the infliximab group; p<0.001). In the infliximab group, the mean change in total modified vdH-S score remained below 0 at the week 54 timepoint. Infliximab-treated patients demonstrated significant improvement in physical function as assessed by HAQ. Significant improvements in health-related quality of life were also demonstrated as measured by the physical and mental component summary scores of the SF-36 in IMPACT 2. Adult psoriasis Intravenous formulation The efficacy of infliximab intravenous formulation was assessed in two multicentre, randomised, double-blind studies: SPIRIT and EXPRESS. Patients in both studies had plaque psoriasis (Body Surface Area [BSA] ≥10% and Psoriasis Area and Severity Index [PASI] score ≥12). The primary endpoint in both studies was the percent of patients who achieved ≥75% improvement in PASI from baseline at week 10. SPIRIT evaluated the efficacy of infliximab induction therapy in 249 patients with plaque psoriasis that had previously received PUVA or systemic therapy. Patients received either 3 or 5 mg/kg infliximab or placebo infusions at weeks 0, 2 and 6. Patients with a PGA score ≥3 were eligible to receive an additional infusion of the same treatment at week 26. In SPIRIT, the proportion of patients achieving PASI 75 at week 10 was 71.7% in the 3 mg/kg infliximab group, 87.9% in the 5 mg/kg infliximab group, and 5.9% in the placebo group (p<0.001). By week 26, twenty weeks after the last induction dose, 30% of patients in the 5 mg/kg group and 13.8% of patients in the 3 mg/kg group were PASI 75 responders. Between weeks 6 and 26, symptoms of psoriasis gradually returned with a median time to disease relapse of >20 weeks. No rebound was observed. EXPRESS evaluated the efficacy of infliximab induction and maintenance therapy in 378 patients with plaque psoriasis. Patients received 5 mg/kg infliximab- or placebo-infusions at weeks 0, 2 and 6 followed by maintenance therapy every 8 weeks through week 22 in the placebo group and through week 46 in the infliximab group. At week 24, the placebo group crossed over to infliximab induction therapy (5 mg/kg) followed by infliximab maintenance therapy (5 mg/kg). Nail psoriasis was assessed using the Nail Psoriasis Severity Index (NAPSI). Prior therapy with PUVA, methotrexate, ciclosporin, or acitretin had been received by 71.4% of patients, although they were not necessarily therapy resistant. Key results are presented in Table 12. In infliximab treated subjects, significant PASI 50 responses were apparent at the first visit (week 2) and PASI 75 responses by the second visit (week 6). Efficacy was similar in the subgroup of patients that were exposed to previous systemic therapies compared to the overall study population. Table 12 Summary of PASI response, PGA response and percent of patients with all nails cleared at Weeks 10, 24 and 50. EXPRESS Placebo → Infliximab Infliximab 5 mg/kg 5 mg/kg (at week 24) Week 10 N 77 301 ≥90% improvement 1 (1.3%) 172 (57.1%) a ≥75% improvement 2 (2.6%) 242 (80.4%) a ≥50% improvement 6 (7.8%) 274 (91.0%) PGA of cleared (0) or minimal (1) 3 (3.9%) 242 (82.9%) ab PGA of cleared (0), minimal (1), or mild 14 (18.2%) 275 (94.2%) ab (2) Week 24 N 77 276 ≥90% improvement 1 (1.3%) 161 (58.3%) a ≥75% improvement 3 (3.9%) 227 (82.2%) a ≥50% improvement 5 (6.5%) 248 (89.9%) PGA of cleared (0) or minimal (1) 2 (2.6%) 203 (73.6%) a PGA of cleared (0), minimal (1), or mild 15 (19.5%) 246 (89.1%) a (2) Week 50 N 68 281 ≥90% improvement 34 (50.0%) 127 (45.2%) ≥75% improvement 52 (76.5%) 170 (60.5%) ≥50% improvement 61 (89.7%) 193 (68.7%) PGA of cleared (0) or minimal (1) 46 (67.6%) 149 (53.0%) PGA of cleared (0), minimal (1), or mild 59 (86.8%) 189 (67.3%) (2) All nails clearedc Week 10 1/65(1.5%) 16/235 (6.8%) Week 24 3/65 (4.6%) 58/223 (26.0%) a Week 50 27/64 (42.2%) 92/226 (40.7%) a p <0.001, for each infliximab treatment group vs. control. b n = 292. c Analysis was based on subjects with nail psoriasis at baseline (81.8% of subjects). Mean baseline NAPSI scores were 4.6 and 4.3 in infliximab and placebo group. Significant improvements from baseline were demonstrated in DLQI (p<0.001) and the physical and mental component scores of the SF 36 (p<0.001 for each component comparison).
Pharmacokinetic Properties
5.2 Pharmacokinetic properties Absorption and distribution Single subcutaneous injections of 120, 180 and 240 mg of infliximab yielded approximately dose proportional increases in the maximum serum concentration (Cmax) and area under the concentration time curve (AUC). The apparent volume of distribution during the terminal phase (mean of 7.3 to 8.8 litres) was not dependent on the administered dose. After single doses of 120, 180 and 240 mg of subcutaneous infliximab administered to healthy subjects, the mean Cmax values were 10.0, 15.1 and 23.1 μg/mL, respectively, and for all doses infliximab could be detected in the serum for at least 12 weeks thereafter. The bioavailability of subcutaneous infliximab, estimated in a population PK model, was 58% (95% CI: 54% - 62%). After administration of infliximab 120 mg subcutaneously every 2 weeks (from Week 6 after 2 doses of intravenous infliximab at Weeks 0 and 2) to patients with active rheumatoid arthritis who were concomitantly treated with MTX, the median (CV%) Ctrough level at Week 22 (steady state) was 12.8 μg/mL (80.1%). After administration of infliximab 120 mg subcutaneously every 2 weeks (from Week 6 after 2 doses of intravenous infliximab at Weeks 0 and 2) to patients with active Crohn’s disease and active ulcerative colitis, the median (CV%) Ctrough level at Week 22 (steady state) was 20.1 µg/mL (48.9%). Based on PK results from clinical studies in patients with active rheumatoid arthritis, active Crohn’s disease and active ulcerative colitis and population PK modelling, Ctrough levels at steady state would be higher after administration of infliximab 120 mg subcutaneous formulation given every 2 weeks compared with infliximab 5 mg/kg intravenous formulation given every 8 weeks. Elimination The elimination pathways for infliximab have not been characterised. Unchanged infliximab was not detected in urine. No major age- or weight-related differences in clearance or volume of distribution were observed in rheumatoid arthritis patients. In studies in healthy subjects, the mean (± SD) apparent clearance of Remsima 120 mg/ml S.C. administered subcutaneously was 19.3 ± 6.9 mL/hr. In the RA patients, the mean (± SD) apparent clearance of Remsima 120 mg/ml S.C. at steady state was 18.8 ± 8.3 mL/hr. In the active Crohn’s disease and active ulcerative colitis patients, the mean (± SD) apparent clearance of Remsima 120 mg/ml S.C. subcutaneous at steady state was 16.1 ± 6.9 mL/hr. The mean terminal half-life ranged from 11.3 days to 13.7 days for 120, 180 and 240 mg of subcutaneous infliximab administered to healthy subjects. Special populations Elderly The pharmacokinetics of infliximab injected via subcutaneous route in elderly patients has not been studied. Hepatic and renal impairment Studies with infliximab have not been performed in patients with liver or renal disease.

פרטי מסגרת הכללה בסל
א. התרופה תינתן לטיפול בחולה הסובל מאחד מאלה: 1. טיפול במחלת קרוהן בדרגת חומרה בינונית עד קשה בחולים שמיצו טיפול קודם – טיפול לא ביולוגי או טיפול ביולוגי;2. ארתריטיס ראומטואידית - אם החולה לא הגיב לטיפול ב-METHOTREXATE והטיפול דרוש לצורך הפחתת הסימנים והתסמינים; הטיפול יינתן בשילוב עם METHOTREXATE ובכפוף לתנאי פסקה ב; 3. דלקת פרקים פסוריאטית קשה אם החולה לא הגיב לטיפול בתרופות methotrexate, salazopyrin ממשפחת ה-DMARDs. הטיפול יינתן בשילוב עם methotrexate; 4. אנקילוזינג ספונדילטיס קשה אם החולה לא הגיב לטיפול קונבנציונלי. במקרה של הוריאנט דמוי אנקילוזינג ספונדיליטיס הקשור בפסוריאזיס, תהיה ההוריה כמו באנקילוזינג ספונדיליטיס ראשונית; 5. פסוריאזיס - בהתקיים כל אלה: א. החולה סובל מאחד מאלה: 1. מחלה מפושטת מעל ל-50% של שטח גוף או PASI מעל 50. 2. נגעים באזורי גוף רגישים - אזורים אלו יכללו פנים, צוואר, קיפולי עור, כפות ידיים, כפות רגליים, אזור הגניטליה והישבן; ב. החולה קיבל שני טיפולים סיסטמיים לפחות בלא שיפור של 50% לפחות ב-PASI לאחר סיום הטיפול בהשוואה לתחילת הטיפול; בהתייחס לחולה העונה על האמור בפסקת משנה (א)(2) - החולה קיבל שני טיפולים סיסטמיים לפחות בלא שיפור משמעותי לאחר סיום הטיפול בהשוואה לתחילת הטיפול; ג. התרופה תינתן על פי מרשם של רופא מומחה בדרמטולוגיה. 6. טיפול במחלת מעי דלקתית מסוג Ulcerative colitis בחולים שמיצו טיפול קודם – טיפול לא ביולוגי או טיפול ביולוגי;ב. הטיפול בתרופה לחולה העונה על תנאי פסקה (א) (2), יינתן בהתקיים כל אלה: 1. קיימת עדות לדלקת פרקים (RA-Rheumatoid Arthritis) פעילה המתבטאת בשלושה מתוך אלה: א. מחלה דלקתית (כולל כאב ונפיחות) בארבעה פרקים ויותר; ב. שקיעת דם או CRP החורגים מהנורמה באופן משמעותי (בהתאם לגיל החולה); ג. שינויים אופייניים ל-RA בצילומי רנטגן של הפרקים הנגועים; ד. פגיעה תפקודית המוגדרת כהגבלה משמעותית בתפקודו היומיומי של החולה ובפעילותו בעבודה. 2. לאחר מיצוי הטיפול בתרופות השייכות למשפחת ה-NSAIDs ובתרופות השייכות למשפחת ה-DMARDs. לעניין זה יוגדר מיצוי הטיפול כהעדר תגובה קלינית לאחר טיפול קו ראשון בתרופות אנטי דלקתיות ממשפחת ה-NSAIDs וטיפול קו שני ב-3 תרופות לפחות ממשפחת ה-DMARDs שאחת מהן מתוטרקסאט, במשך 3 חודשים רצופים לפחות. 3. הטיפול יינתן באישור רופא מומחה בראומטולוגיה.
מסגרת הכללה בסל
התוויות הכלולות במסגרת הסל
| התוויה | תאריך הכללה | תחום קליני | Class Effect | מצב מחלה |
|---|---|---|---|---|
| פסוריאזיס, בהתקיים כל התנאים האלה: (1) החולה סובל מאחד מאלה: (א) מחלה מפושטת מעל ל-50% של שטח גוף או PASI מעל 50; (ב) נגעים באזורי גוף רגישים; (2) החולה קיבל שני טיפולים סיסטמיים לפחות בלא שיפור של 50% לפחות ב-PASI לאחר סיום הטיפול בהשוואה לתחילת הטיפול; בהתייחס לחולה העונה על האמור בפסקת משנה (1)(ב) – החולה קיבל שני טיפולים סיסטמיים לפחות בלא שיפור משמעותי לאחר סיום הטיפול בהשוואה לתחילת הטיפול; (3) התרופה תינתן על פי מרשם של רופא מומחה בדרמטולוגיה | 03/01/2010 | עור ומין | ADALIMUMAB, IXEKIZUMAB, CERTOLIZUMAB PEGOL, USTEKINUMAB, SECUKINUMAB, TILDRAKIZUMAB, GUSELKUMAB, ETANERCEPT, INFLIXIMAB | Psoriasis |
| טיפול במחלת מעי דלקתית מסוג Ulcerative colitis לאחר מיצוי כל קווי טיפול תרופתיים קיימים | 01/03/2008 | גסטרואנטרולוגיה | Ulcerative colitis | |
| פסוריאזיס בהתקיים כל התנאים האלה א. החולה סובל מפסוריאזיס מפושטת מעל ל-50% של שטח גוף או PASI מעל 50. ב. הטיפול עם התכשיר יינתן לחולים אשר קיבלו לפחות שני טיפולים סיסטמיים ללא שיפור של 50% לפחות ב-PASI לאחר סיום הטיפול בהשוואה לתחילת הטיפול. ג. לא יעשה שימוש בשתי תרופות מקבוצה זו (התרופות בקלאס אפקט) בתוך 12 חודשים, אלא אם קיימת אי סבילות או תופעות לוואי לתרופה המחייבות זאת. ד. התרופה תינתן על פי מרשם של רופא מומחה בדרמטולוגיה. | 15/05/2006 | עור ומין | EFALIZUMAB, ALEFACEPT, ETANERCEPT, INFLIXIMAB | Psoriasis |
| דלקת מפרקים ראומטואידית אם החולה לא הגיב לטיפול ב-Methtorexate והטיפול דרוש לצורך הפחתת הסימנים והתסמינים; הטיפול יינתן בשילוב Methotrexate בהתקיים כל התנאים הבאים 1. קיימת עדות לדלקת מפרקים מסוג Rheumatoid arthritis פעילה המתבטאת בשלושה מתוך אלה: א. מחלה דלקתית בארבעה מפרקים ויותר ב. שקיעת דם או C reactive protein מעל הנורמה ג. שינויים אופייניים לדלקת מפרקים ראומטואידית במפרקים הנגועים ד. פגיעה תפקודית לאחר מיצוי הטיפול בתרופות השייכות למשפחת ה-NSAIDs (Non steroidal anti inflammatory drugs) ובתרופות השייכות למשפחת DMARDs (Disease modifying antirheumatic drugs). 2. התרופה תינתן על פי המלצה של רופא מומחה בראומטולוגיה. | 15/04/2005 | ראומטולוגיה | Rheumatoid arthritis | |
| אנקילוזינג ספונדיליטיס קשה – אם החולה לא הגיב לטיפול קונבנציונלי. הטיפול יינתן בשילוב עם Methotrexate, למעט במקרה של הורית נגד לתרופה זאת. התרופה תינתן על פי המלצה של רופא מומחה בראומטולוגיה. | 15/04/2005 | ראומטולוגיה | Ankylosing spondylitis | |
| דלקת מפרקים פסוריאטית קשה – אם החולה לא הגיב לטיפול בתרופות Salazopyrine, Methotrexate ממשפחת ה-DMARDs. הטיפול יינתן בשילוב עם Methotrexate. (במקרה של הוריאנט דמוי Ankylosing spondylitis הקשור בפסוריאזיס תהיה ההוריה כמו ב-Ankylosing spondylitis ראשוני). התרופה תינתן על פי המלצה של רופא מומחה בראומטולוגיה. | 15/04/2005 | ראומטולוגיה | Psoriatic arthritis | |
| ארתריטיס ראומטואידית – אם החולה לא הגיב לטיפול ב-METHOTREXATE והטיפול דרוש לצורך הפחתת הסימנים והתסמינים; הטיפול ינתן בשילוב עם METHOTREXATE. התוויות למתן הטיפול: הטיפול בחוסמי TNF מיועד רק לחולי ארתריטיס ראומטואידית פעילה, בהתקיים כל התנאים שיפורטו להלן: 1. עדות קלינית מעבדתית, רנטגנית ותפקודית לדלקת פרקים (RA) פעילה (לפחות 3 מתוך 4 הבאים): א. פגיעה קלינית מוגדרת כמחלה דלקתית (כולל כאב ונפיחות) במספר פרקים בו זמנית (לפחות 4 פרקים) ב. עדות מעבדתית למחלה דלקתית פעילה מוגדרת – שקיעת דם ו/או CRP החוגרים מהנורמה באופן משמעותי (בהתאם לגיל החולה). ג. עדות רנטגנית מוגדרת כשינויים אופייניים ל-RA בצילומי הרנטגן של הפרקים הנגועים. הערה: קיום אנקילוזות במרבית הפרקים (כביטוי לשלב הקליני הסופי של המחלה), אינו מהווה הוריה למתן הטיפול. ד. פגיעה תפקודית עקב מחלה פעילה מוגדרת כהגבלה משמעותית בתפקודו היומיומי של החוולה ובפעילותו בעבודה. 2. ניסיון טיפולי קודם בתרופות הבאות: א. טיפול קו ראשון בתרופות אנטי דלקתיות מסוג NSAID ב. טיפול קו שני ב-3 תרופות לפחות מקבוצת ה-DMARD שאחת מהן מתוטרקסט, במשך 3 חודשים רצופים לפחות. קבוצת התרופות DMARD (Disease modifying antirheumatic drugs) כוללת: מתוטרקסט, מלחי זהב, אנטימלריאלים, דיפניצילאמין, סולפהסלזין, אזאתיופרין, מינוציקלין. כשלון טיפולי יוגדר כהעדר תגובה קלינית לאחר טיפול של 3 חודשים רצופים לפחות בטיפול של 3 תרופות מקבוצת ה-DMARD, שאחת מהן מתוטרקסט. כשלון טיפול ב-Infliximab יוגדר כהעדר תגובה קלינית לאחר טיפול ב-4 מנות של התכשיר במתן תוך ורידי. הנחיות למתן הטיפול חוסמי TNF יינתנו כטיפול קו שלישי רק לאחר כשלון טיפולי בתרופות קו שני ולפי ההוריות הבאות: 1. הטיפול ב-Infliximab ב-Adult RA יינתן בשילוב עם Methotrexate. 2. התרופה Etanercept תינתן: א. לטיפול במחלת ארתריטיס כרונית בצעירים (Juvenile chronic arthritis) לאחר כשלון טיפולי במתוטרקסט. ב. לטיפול ב-Adult RA לאחר כשלון טיפולי ב-DMARD כמתואר לעיל, ולאחר כשלון טיפולי ב-Infliximab. שמירת רצף טיפול בחוסמי TNF ישמר הרצף הטיפולי במתן חוסמי TNF בחולים אשר ענו על ההתוויות הקליניות המוגדרות לפני תחילת הטיפול, באותה תרופה בה הותחל הטיפול לפני 01.01.02 וקיימת הוכחה ליעילות הטיפול. אישור מתן הטיפול הטיפול בחוסמי TNF בחולי ארתריטיס ראומטואידית יינתן באישור וועדה בראשות רופא מומחה בראומטולוגיה. | 01/03/2002 | ראומטולוגיה | Rheumatoid arthritis | |
| בחולה הסובל מאחד מאלה: (א) מחלת קרוהן פעילה בינונית עד חמורה, לצורך הקלת הסימנים והתסמינים. (ב) מחלת קרוהן מסוג fistulizing - לצורך הפחתת מספר הפיסטולות האנטרו-עוריות המנקזות (draining enterocutaneous fistulas). | 16/01/2000 | גסטרואנטרולוגיה | Crohn's disease |
שימוש לפי פנקס קופ''ח כללית 1994
לא צוין
תאריך הכללה מקורי בסל
16/01/2000
הגבלות
תרופה מוגבלת לרישום ע'י רופא מומחה או הגבלה אחרת
מידע נוסף
עלון מידע לצרכן
18.05.22 - עלון לצרכן אנגלית 18.05.22 - עלון לצרכן עברית 18.05.22 - עלון לצרכן ערבית 20.12.21 - עלון לצרכן אנגלית 14.06.22 - עלון לצרכן עברית 20.12.21 - עלון לצרכן ערבית 12.10.22 - עלון לצרכן אנגלית 12.10.22 - עלון לצרכן עברית 12.10.22 - עלון לצרכן ערבית 29.05.23 - עלון לצרכן אנגלית 29.05.23 - עלון לצרכן עברית 29.05.23 - עלון לצרכן ערבית 08.06.23 - עלון לצרכן אנגלית 08.06.23 - עלון לצרכן עברית 08.06.23 - עלון לצרכן ערבית 15.08.23 - עלון לצרכן אנגלית 15.08.23 - עלון לצרכן עברית 15.08.23 - עלון לצרכן ערבית 24.10.23 - עלון לצרכן עברית 25.10.23 - עלון לצרכן עברית 20.12.23 - עלון לצרכן אנגלית 20.12.23 - עלון לצרכן עברית 20.12.23 - עלון לצרכן ערבית 20.12.21 - החמרה לעלון 10.05.23 - החמרה לעלון 19.10.23 - החמרה לעלוןלתרופה במאגר משרד הבריאות
רמסימה 120 מ"ג/מ"ל תת-עורי
